

2019/03/20
生体内の細胞1個ずつについて遺伝子の働き具合を調べる手法を開発
切断されたコケの細胞の初期化、再生に伴う個別の変化をつきとめる
~ 生き物の形作りや刺激応答の仕組み解明に期待 ~
【概要】
奈良先端科学技術大学院大学(学長:横矢 直和)研究推進機構ヒューマノフィリックイノベーション科学技術研究推進事業の 久保 稔 特任准教授、先端科学技術研究科バイオサイエンス領域の 出村 拓 教授、佐野 亮輔 研究員と、金沢大学、基礎生物学研究所、総合研究大学院大学、広島工科大学、ドイツ フライブルク大学などの国際共同研究グループは、生体内にある個別の細胞について遺伝子の働き具合を調べるための新たな手法の開発に成功しました。マイクロキャピラリー1*という実験器具を用いて生きた組織から1個の細胞を単離する技術と、超高速の解析装置である次世代シーケンサー2*を利用するさいに遺伝子の活性化の指標となるメッセンジャーRNA(mRNA)3*のコピー数を数えるRNA-seq4*という定量法を組み合わせた手法(1cell-DGE)です。さらにこの手法により、モデル植物であるヒメツリガネゴケ5*から植物細胞の初期化、再生に関わる6382個の発現変動遺伝子7*を見出し、葉細胞から幹細胞にリプログラミング6*する過程で幹細胞になる細胞と変化しない細胞の集団の特定に初めて成功しました。
これまでの方法では、個別の細胞について遺伝子の働き具合を調べるときに、細胞をバラバラにすることが前提となっており、それが不可能な生物組織の細胞では、解析ができませんでした。また、細胞をバラバラにすることで隣接する細胞との位置情報が失われるため、他の細胞との関係性を調べることも困難でした。
しかし、今回開発した1cell-DGE法を用いることで、あらゆる生物組織の細胞を分離して、隣接する細胞の位置情報まで利用した1細胞遺伝子発現解析が可能となります。多くの細胞の集まりである組織や器官、個体において、どのように個々の細胞がお互いに協力して、組織、器官や個体を作り上げるのか、また、環境からの刺激に対して組織内の細胞がどのように応答するのかという生き物の形作りや刺激応答の基本的な問題に対して新たな知見を得ることが期待されます。
この研究成果は、平成31年3月15日に英国の国際学術雑誌「Nucleic Acids Research」オンライン版に掲載されました。
つきましては、関係資料を配付いたしますので、取材の方、よろしくお願いいたします。
【ご連絡事項】
- 本件につきましては、奈良先端科学技術大学院大学から奈良県文化教育記者クラブをメインとし、学研都市記者クラブ、大阪科学・大学記者クラブへ同時にご連絡しております。
- 取材希望がございましたら、恐れ入りますが下記までご連絡願います。
- プレスリリースに関する問い合わせ先
奈良先端科学技術大学院大学 研究推進機構
ヒューマノフィリックイノベーション科学技術研究推進事業 特任准教授 久保 稔
TEL :0743-72-6158
E-mail:[email protected]
【論文情報】
タイトル:
Single-cell transcriptome analysis of Physcomitrella leaf cells during reprogramming using microcapillary manipulation
著者:
Minoru Kubo1,*, Tomoaki Nishiyama2, Yosuke Tamada3,4, Ryosuke Sano5, Masaki Ishikawa3,4, Takashi Murata3,4, Akihiro Imai6, Daniel Lang7, Taku Demura5, Ralf Reski7,8,* and Mitsuyasu Hasebe3,4,*
所属:
1Institute for Research Initiative, Nara Institute of Science and Technology(奈良先端科学技術大学院大学・研究推進機構)
2Advanced Science Research Center, Kanazawa University(金沢大学)
3National Institute for Basic Biology(基礎生物学研究所)
4School of Life Science, The Graduate University for Advanced Studies(総合研究大学院大学)
5Graduate School of Science and Technology, Nara Institute of Science and Technology(奈良先端科学技術大学院大学・先端科学技術研究科)
6Faculty of Life Sciences, Hiroshima Institute of Technology(広島工科大学)
7Plant Biotechnology, Faculty of Biology, University of Freiburg(ドイツ フライブルク大学)
8Signaling Research Centres BIOSS and CIBSS, University of Freiburg(ドイツ フライブルク大学)
*共同責任著者
掲載誌:
Nucleic Acids Research
DOI:
10.1093/nar/gkz181
【背景】
次世代シーケンサー2*を用いて遺伝子の働き具合を調べるためには、遺伝子DNAのコピーであるメッセンジャーRNA(mRNA)3*のコピー数を数える、RNA-seq解析4*が広く行われています。しかし、この解析に必要なmRNAを集めるには、数十個から数万個の細胞を集め、それをすりつぶしてmRNAを取り出す必要があります。このため、得られたmRNAのコピー数は、集められた細胞数あたりの平均値にすぎず、個々の違いはわかりませんでした。また、様々な刺激への応答や、発生の段階で細胞が分化していく過程を調べるためにも、ある決まった時間ごとに細胞をサンプリングする必要がありますが、たとえ同調的に細胞の分化の過程が進んでいても、その中には分化の進み具合が異なる細胞が常に混入しており、そのためmRNAの数は、その時サンプリングした細胞あたりの平均値にすぎませんでした。
近年、1個の細胞内にある遺伝子のコピー数を検出する1細胞RNA-seq(scRNA-seq)解析法8*の技術が急速に発展してきました。この解析法により、これまで同じグループに属する細胞の集まりの中にも、細胞それぞれに個性があることや、ごく少ない数の珍しい細胞が含まれることが明らかになってきました。さらに、細胞分化過程においては遺伝子のコピー数が似ている1個1個の細胞を順番に並べることで、サンプリングしていないタイミングの細胞の分化状態をも詳細に予測することができるようになりました。
ところが、このscRNA-seqを行うためには、(1)細胞をバラバラにすること、(2)1細胞内の少ないmRNAを次世代シーケンサーで解析できるまでに増やすこと、が必須でした。そのため、細胞壁で強固に結合した植物細胞においては、細胞をバラバラにすることが困難であり、多くの場合、scRNA-seq解析を行うことは不可能でした。
【方法と結果】
そこで著者らは、細胞をバラバラにせず組織内にあるままの状態で、生きた植物細胞内にマイクロキャピラリー1*を突き刺して核を抽出し、そのmRNAを利用した次世代シーケンサー用のライブラリーを作製する手法(1cell-DGE)を確立しました(図1)。1cell-DGEでは、mRNAをDNAに変換して計測しますが、その際、バーコードのようなランダムな塩基配列(Unique Molecule Identifier: UMI9*)を目印として付け加えます。これで、次世代シーケンサーによる解析に必要なDNA量を確保するため、PCR10*でという方法で大量増殖しても、UMIという目印を手掛かりにして最初にあったmRNAの数がわかるようになっています。
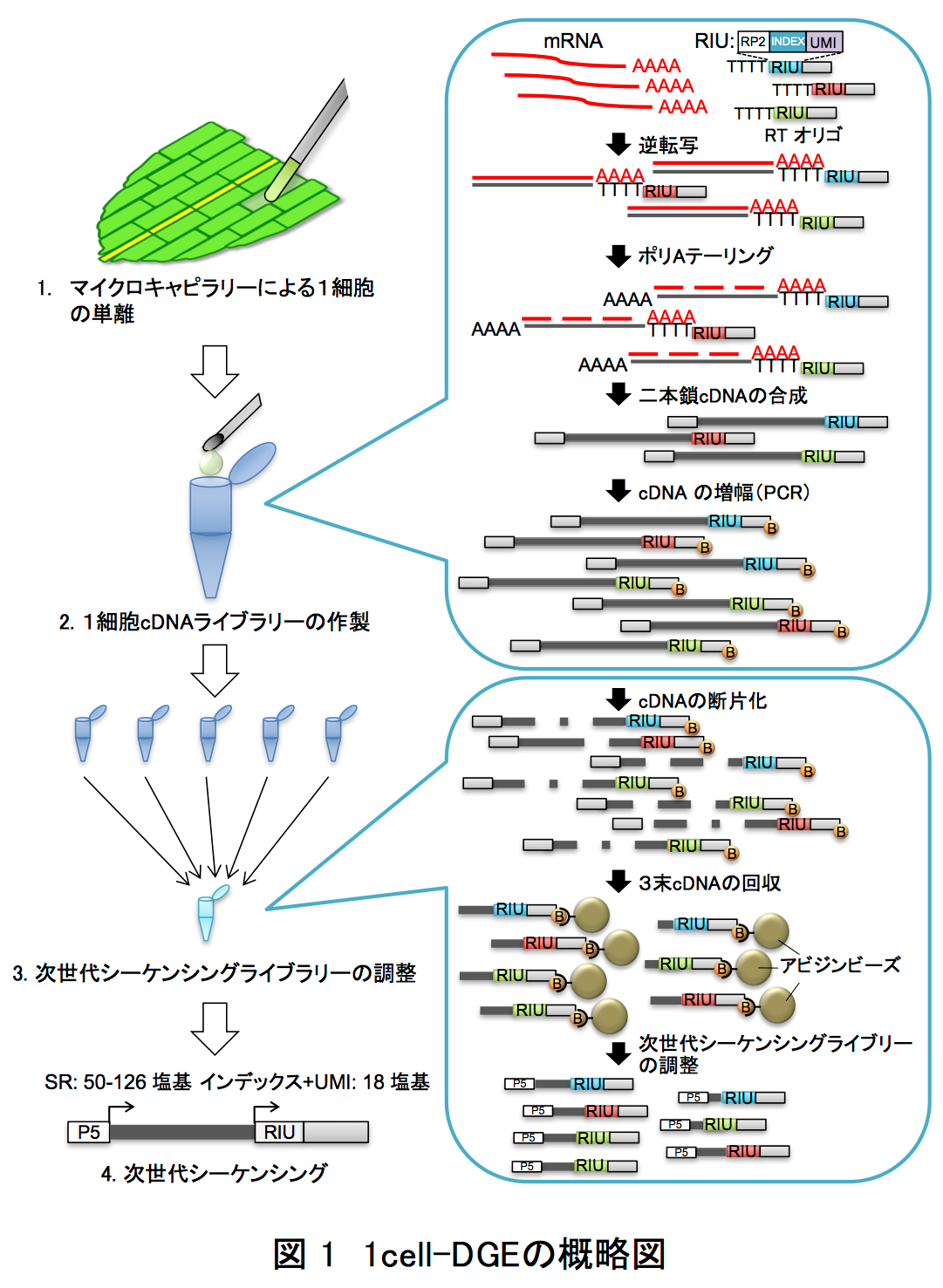

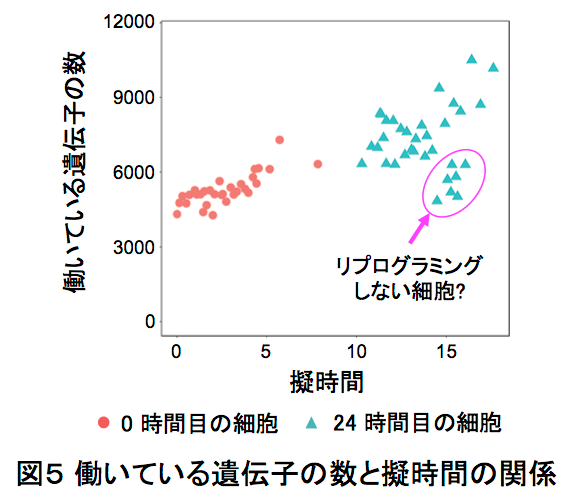
この1cell-DGEを用いて、ヒメツリガネゴケ5*の切断した葉のリプログラミング6*時におけるscRNA-seq解析を行いました。ヒメツリガネゴケの切断された面にある幾つかの葉細胞は、先端に幹細胞を持つ原糸体細胞11*にリプログラミングします(図2)。葉切断後、0時間目と24時間目の切り口に面した葉細胞をそれぞれ31個と34個、マイクロキャピラリーを用いて核部分を単離した1cell-DGEを行いました。その結果、6382個もの発現変動遺伝子7*の検出に成功しました(図3)。特に24時間目では切断した葉全体を用いたRNA-seqに比べて、6倍以上もの発現変動遺伝子が見いだしました(図4)。この発現変動遺伝子のRNAのコピー数をもとにリプログラミング過程の進行の指標になる擬時間(Pseudotime)12*を計算し、働いている遺伝子の数との相関を調べると、擬時間が進むにつれ、0時間目の細胞は働いている遺伝子の数が増えているのに対し、24時間目の細胞では擬時間の終盤において、働いている遺伝子が多い細胞と少ない細胞のグループに分かれることが観察されました(図5)。このことから、リプログラミングが完了していない24時間目の細胞では、リプログラミングが進んでいる細胞と、すでにリプログラミングしない細胞が存在することが示唆されました。


【今後の展開】
この1cell-DGEを用いることで、(1)細胞をバラバラにすることができない生物組織、(2)隣接する細胞の位置情報、を利用したscRNA-seq解析が可能となり、多くの細胞の集まりである組織や器官、個体において、どのように個々の細胞が秩序を保ちつつ、組織、器官や個体を作り上げるのか、また、環境からの刺激に対して個々の細胞はどのように応答するのかという問題に対して新たな知見を得ることが期待されます。
【研究プロジェクト】
本研究は、日本学術振興会「頭脳循環を活性化する若手研究者海外派遣プログラム」、科研費「新学術領域 公募研究(JP26113514)」,奈良先端科学技術大学院大学 「ヒューマノフィリックイノベーション科学技術研究推進事業」、「多元ビッグデータ解析に基づく知の創出研究拠点事業」、「戦略的国際学術論文校正・掲載支援プロジェクト」、Excellence Initiative of the German Federal and States Governments(EXC294)の資金的支援を受けて実施されました。
【用語解説】
- マイクロキャピラリー:先端が細く尖った中空のガラス管。
- 次世代シーケンサー:一度に数千万個から数億個の短いDNA断片の塩基配列を解読する装置。
- メッセンジャーRNA(mRNA):DNA上にある遺伝子部分を鋳型として転写(コピー)されるRNA。これを元にタンパクが翻訳されるため、多くの場合、mRNAのコピー数が多いほどよく働いている遺伝子とみなされる。
- RNA-seq:次世代シーケンサーを用いて行うmRNAのコピー数の定量法。mRNAをDNAに変換してから塩基配列を解読し(リードと呼ばれる)、それらが各遺伝子に何リードあるか数えることでmRNAのコピー数を数える。
- ヒメツリガネゴケ:基部陸上植物であるコケ植物セン類に属するモデル植物の一種。解読された全ゲノム情報によって32926遺伝子を持つことがわかっている。
- リプログラミング:体細胞が幹細胞に変化する生物に共通した現象。
- 発現変動遺伝子:mRNAのコピー数がサンプル間で有意に変わっている遺伝子。この場合、0時間目と24時間目でmRNAのコピー数が有意に異なる遺伝子を指す。
- scRNA-seq:1細胞用に開発されたRNA-seqの方法。1細胞遺伝子発現解析ともいう。
- Unique Molecular Identifier(UMI):次世代シーケンサーによって識別されるランダムな6から10個の塩基配列。1cell-DGEの場合、mRNAからDNAの変換時に10個の塩基が付けられる。
- PCR:ポリメラーゼ連鎖反応。耐熱性DNAポリメラーゼと短いDNA(プライマー)を用いて、鋳型となるDNAを増やす方法。
- 原糸体:コケ植物に見られる糸状に細胞が連なった組織。先端に幹細胞としての原糸体頂端細胞がある。
- 擬時間(Pseudotime):発現変動遺伝子の情報を元に計算された相対的な細胞の分化・遷移状態を示す指標。
【本プレスリリースに関するお問い合わせ先】
奈良先端科学技術大学院大学 研究推進機構
ヒューマノフィリックイノベーション科学技術研究推進事業
久保 稔(くぼ みのる)特任准教授
E-mail:[email protected]


